
Scott M. Palmer,1,2 Megan L. Neely,1,2 John A. Belperio,3 Shaun Bender,4 Daniel A. Culver,5 Joao de Andrade,6 Emily C. O’Brien,1 Jesse Roman,7 Laurie D. Snyder,1,2 Jamie L. Todd,1,2 Timothy P.M. Whelan,8 Thomas B. Leonard,4 Craig S. Conoscenti4 on behalf of the ILD-PRO Registry investigators
1Duke Clinical Research Institute, Durham, North Carolina, USA; 2Duke University Medical Center, Durham, North Carolina, USA; 3David Geffen School of Medicine at UCLA, Los Angeles, California, USA; 4Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, Connecticut, USA; 5Cleveland Clinic, Cleveland, Ohio, USA; 6Vanderbilt University, Nashville, Tennessee, USA; 7Jane and Leonard Korman Respiratory Institute, Philadelphia, Pennsylvania, USA; 8Medical University of South Carolina, Charleston, South Carolina, USA.
Introduction
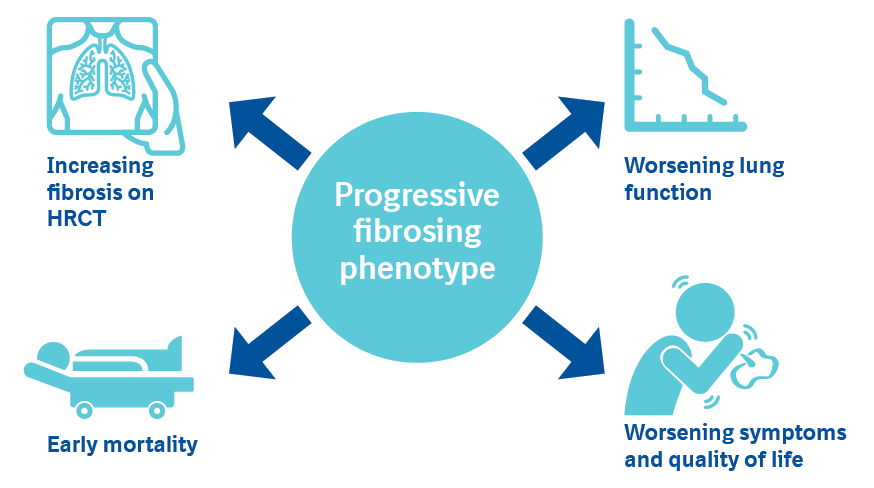
- Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing ILD that is always progressive. A subset of patients with other chronic fibrosing ILDs also develop a progressive phenotype characterized by increasing fibrosis on HRCT; worsening lung function, symptoms and quality of life; and early mortality.1,2
- In 2018, the Idiopathic Pulmonary Fibrosis Prospective Outcomes (IPF-PRO) Registry,3 an observational US registry involving 1002 patients with IPF, was expanded to include an additional arm comprising patients with non-IPF chronic progressive fibrosing ILDs to form the IPF-PRO/ILD-PRO Registry.